GALACTOSEMIA – Definition, Diagnosis and Treatment
DEFINITION
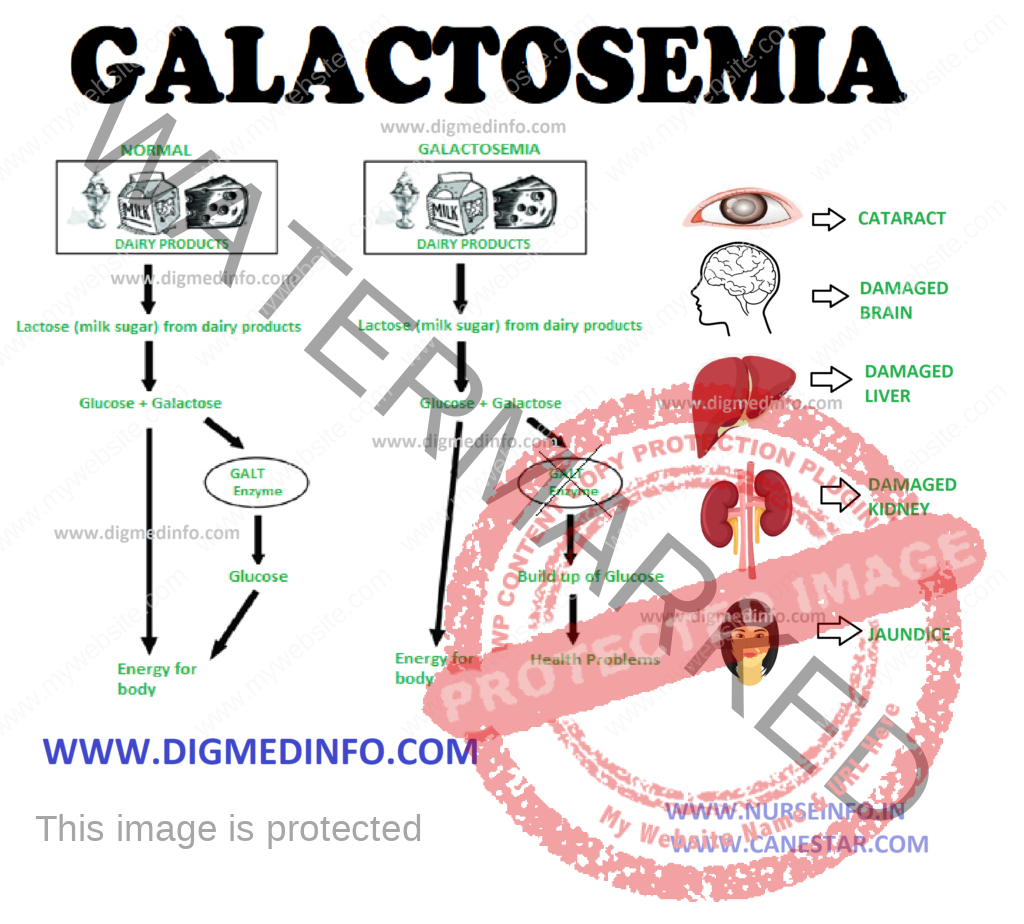
Normally lactose present in milk is converted into glucose and galactose by intestinal lactase. Galactose is absorbed and converted to glucose in the liver. Conversion into glucose is defective in galactosemia and hence the concentration of galactose-1-phosphate and galactose increase in blood and tissues such as liver, brain, kidneys, intestines, and lens.
Galactosemia is transmitted as an autosomal recessive trait. In the fully developed form, classical galactosemia presents with cataract, mental retardation, hepatic cirrhosis, and death in early life. The enzyme galactose-1-phosphate uridyl transferase is deficient. The severe form manifests within a few days of birth with intolerance to milk, vomiting, refusal to feed, and failure to thrive. Jaundice, hepatomegaly, and hepatic dysfunction develop early. Cataracts develop within weeks or months. As the child grows up, mental deficiency becomes evident. Recurrent bacterial infections (E. coli) are common and may be fatal.
In the atypical form of galactosemia, the enzyme galactokinase is deficient. Galactokinase is required to convert galactose into galactose-1-phosphate and in its absence galactose accumulates in blood and tissues. The only complication in this disorder is cataract formation.
Diagnosis
Urine shows galactose. Diagnosis is established by demonstrating the deficiency of galactose-1-phosphate uridyl transferase in erythrocytes.
TREATMENT
Treatment consists of dietary measures to avoid milk and milk products, and these results in dramatic improvement. Even in advanced cases dietary management leads to regression of symptoms except the cataract.